В тему приглашаются знатоки квантовой механики, физхимики и химики-органики (вторых и третьих тут наверно нет, ну да ладно).
Я занимаюсь квантовой химией, и у меня давно были мысли сделать реализацию молекулярной механики. Говорят что современная ММ иногда неплохо работает, например позволяет находить конформации органических молекул. И создалось ощущение, что я могу придумать некий новый подход.
Общая идея у меня такая. Мы проводим с квантовохимическим методом, например DFT, расчёт молекулы, и далее используем результаты расчёта как выборку для обучения функционала молекулярной механики, или как это назвать, ну в общем мы подгоняем параметры нашего ММ функционала так, чтобы получилось наибольшее согласие с результатами DFT расчёта. И далее полученный кастомный ММ функционал мы используем, чтобы посчитать более сложную задачу, для которой исходный DFT метод слишком дорогостоящий. Как пример: мы посчитали на DFT колебательный спектр (вторые производные по энергии от координат атовов), далее загоняем силовое поле и другие параметры для подгонки ММ функционала, и далее полученный ММ функционал используем для расчёта ангармонического силового поля, т.е. третьих производных (это позволяет рассчитать более точную колебательную энтропию или обертоны в колебательном спектре).
Я пока не очень знаю, насколько это будет работать, но мне кажется есть шанс, что получится что-то грандиозное.
Так вот. Для такого подхода нужно реализовывать ММ модель такую, чтобы ММ расчёт в ней был скорее не “хороший”, а “неэмпиричный”. Т.е. эта ММ модель должна основываться на каких-то универсальных, фундаментальных принципах, тогда в общем случае подгонка параметров ММ функционала будет работать неплохо. Что я имею в виду под фундаментальными принципами? Ну например стерическое отталкивание. Очевидно, можно считать универсальным принципом, что несвязанные атомы обычно отталкиваются.
Поясню, что методы квантовой химии делятся на неэмпирические (ab initio) и эмпирические подходы. Ab initio подход означает, что мы проводим расчёт свойств молекулы, решая уравнение Шредингера; мы при этом используем только знания квантовой механики и универсальных физических постоянных. Но ab initio подход часто слишком дорогостоящий, т.е. ресурсов компьютера не хватает для его использования; и тогда мы вводим разные приближения, плюс подгончные параметры, которые подгоняем под данные эксперимента для изучаемых молекул. Это называется эмпирическим подходом, и он менее универсален — для молекул, по которым мы делали подгонку, он может и будет работать, а для непохожих систем уже нет.
Другим универсальным принципом может служить потенциал Леннарда-Джонса. Насколько универсальна его формула?
Может быть я немного путаю, я недавно думал что потенциал Леннарда-Джонса описывает поверхность потенциальной энергии (ППЭ) двухатомных молекул; если нет, каким потенциалом описывается эта ППЭ и насколько он универсален?
Извиняюсь если написанное выше многим непонятно, но я могу провести ликбез и разъяснить все эти моменты, если интересно. И далее я напишу конкретнее про мои идеи.
"Ты должен сделать добро из зла, потому что его больше не из чего сделать". АБ Стругацкие.
Возможно тут не знают что такое молекулярная механика; кратко, это подход, когда молекула описывается набором координат атомов, и программа считает потенциальную энергию как функцию от этих координат; эта энергия является суммой составляющих по связям в молекуле и по углам, может по ещё каким-то параметрам. Если можно рассчитать потенциальную энергию для любого набора координат, можно подогнать координаты чтобы довести её до минимума (оптимизация геометрии), рассчитать первые, вторые и так далее производные энергии по координатам — это называется силовым полем и оно нужно например чтобы посчитать ИК спектр молекулы,
"Ты должен сделать добро из зла, потому что его больше не из чего сделать". АБ Стругацкие.
Re[2]: Реализация ab initio подхода в молекулярной механике
Здравствуйте, Khimik, Вы писали:
K>Возможно тут не знают что такое молекулярная механика; кратко, это подход, когда молекула описывается набором координат атомов, и программа считает потенциальную энергию как функцию от этих координат; эта энергия является суммой составляющих по связям в молекуле и по углам, может по ещё каким-то параметрам. Если можно рассчитать потенциальную энергию для любого набора координат, можно подогнать координаты чтобы довести её до минимума (оптимизация геометрии), рассчитать первые, вторые и так далее производные энергии по координатам — это называется силовым полем и оно нужно например чтобы посчитать ИК спектр молекулы,
Наиболее конструктивный подход, имхо:
1. Погуглить недавние статьи на тему моделирования молекулярной механики.
2. Связаться с их авторами, предложить коллаборацию.
Я думаю, что на RSDN нет людей, кто занимался бы этой тематикой; поэтому шансы найти полезных единомышленников слишком низки.
Уйдемте отсюда, Румата! У вас слишком богатые погреба.
Re: Реализация ab initio подхода в молекулярной механике
Мне тут предложили поизучать программу USPEX, нечто в таком же духе, только круче.
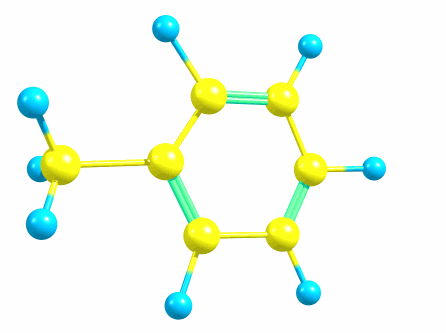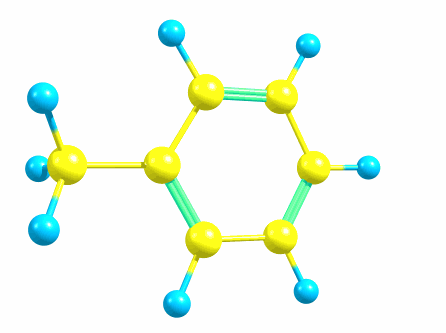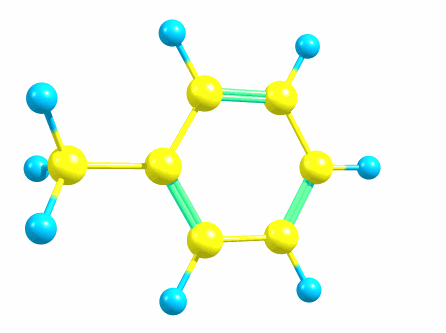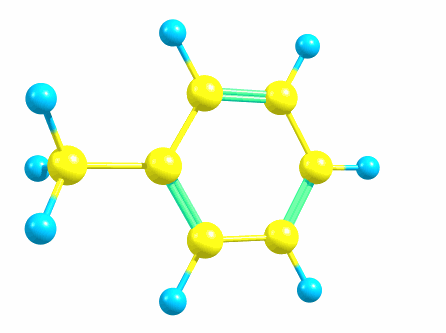
Хотелось бы немного рассказать про квантовую химию. Предположим, нам надо рассчитать свойства молекулы толуола:
Квантовохимический расчёт подразумевает, что мы даём программе координаты атомов, точнее ядер, и число электронов в системе. С точки зрения программы, молекула это
Далее программа решает уравнение Шредингера, причём именно электронное, и находит полную энергию системы. Эта энергия включает в себя потенциальную энергию отталкивания ядер (предполагается что ядра неподвижны) и энергию электронов.
Далее программа меняет координаты атомов и повторяет расчёт так, чтобы полная энергия была как можно ниже. Это называется оптимизация геометрии; предполагается, что эта структура с минимальной энергией будет как бы "истинной" структурой считаемой молекулы (равновесная геометрия). Вот так выглядит типичная оптимизация геометрии:
Далее программа может поизучать, как потенциальная энергия молекулы зависит от её геометрии, от координат ядер (это называется поверхность потенциальной энергии). Обычно рассчитывают ППЭ в гармоническом приближении — для равновесной геометрии считают вторые производные энергии по координатам атомов. В гармоническом приближении можно решить ядерное уравнение Шредингера, и получить колебательный спектр (инфракрасный или рамановский). Иногда считают и третьи производные, это дорогостоящая задача но спектр получится лучше.
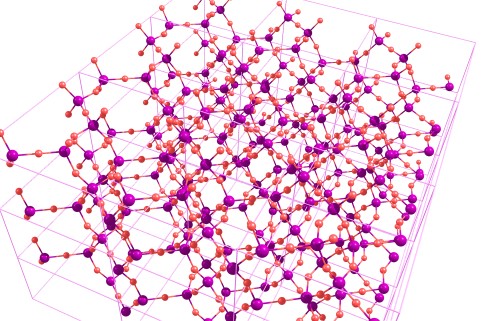
Для кристаллов квантовохимический расчёт включает использование периодических граничных условий (PBC): считается как бы бесконечный континуум, состоящий из одинаковых ячеек:
Продолжение следует.
"Ты должен сделать добро из зла, потому что его больше не из чего сделать". АБ Стругацкие.
Re: Реализация ab initio подхода в молекулярной механике
Сейчас я пытаюсь понять, что такое делокализация "с точки зрения уравнения Шрёдингера".
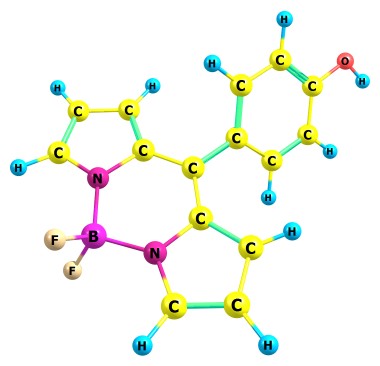
Для некоторых органических молекул трудно понять, как описать внутримолекулярные взаимодействия в рамках учебника органики. Вот например молекула бодипая:
Здесь есть две связи B-N, у которых непонятно какой порядок связи. Мне показалось, химик мог бы рассуждать так: одна связь одинарная, а вторая донорно-акцепторная (или может ионная), и делокализация приводит к их перемешиванию.
В методе молекулярной механики, мы задаём набор координат атомов и типы связей между ними, и считаем энергию как сумму по отдельным связям. Так вот я бы поместил эти две связи в единую группу, и энергия каждой тогда будет считаться как сумма по двум вариантам типов связи.
Для бензола, с таким подходом можно считать, что есть три одинарные связи C-C и три двойные, и все шесть помещаем в единую группу.
В методе молекулярной механики энергия каждой связи, точнее её вклад в полную энергию, считается как функция от длины связи — обычно как парабола (т.е. минимум при R, соответствующем экспериментальной длине связи в двухатомной молекуле). А длина связи в многоатомной молекуле считается по декартовым координатам атомов (формула Пифагора). Надеюсь кому-то стало понятнее.
"Ты должен сделать добро из зла, потому что его больше не из чего сделать". АБ Стругацкие.